Markdown格式的Samtools文章
一、文章标题
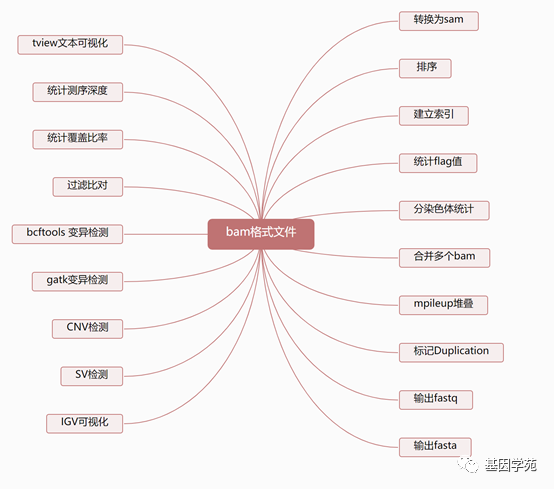
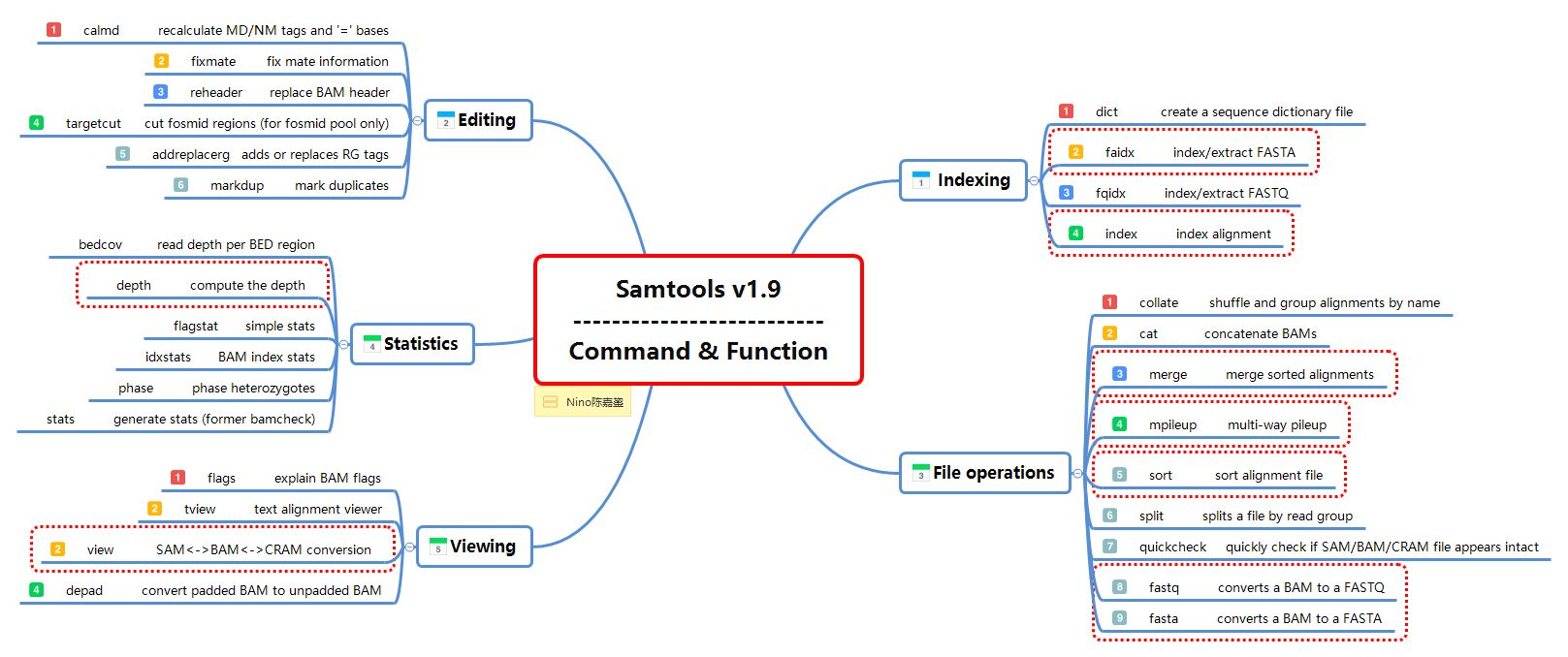
Samtools简介

二、文章内容
Samtools是一款开源的生物信息学软件,用于处理和分析高通量测序数据(如基因组学和转录组学数据)。在大数据的生物信息学研究中,Samtools起到了至关重要的作用。下面我们将从其背景、功能、使用场景和总结等方面,详细介绍Samtools。
- Samtools的背景
Samtools最初由Heng Li和Brenton Garrison等人开发,并由开源社区持续维护和更新。它是在生物信息学领域广泛使用的工具之一,主要用于处理和分析测序数据中的序列比对文件(如SAM、BAM和CRAM格式)。
- Samtools的功能
Samtools提供了许多强大的功能,可以处理和操作序列比对文件。这些功能包括:排序、去重复、转化文件格式等。通过Samtools的操作,研究者们能够方便地对高通量测序数据进行处理和分析。
(1)排序功能:Samtools可以将序列比对文件按照染色体位置进行排序,使得后续的分析更加准确和高效。
(2)去重复功能:通过对高通量测序数据进行去重复处理,Samtools能够减少冗余的数据量,降低存储成本和分析复杂度。
(3)转化文件格式:Samtools能够转化序列比对文件的格式,如将SAM格式转化为BAM或CRAM格式,以适应不同的分析需求。
- Samtools的使用场景
Samtools在生物信息学研究中有着广泛的应用场景。例如,在基因组学研究中,研究者们可以使用Samtools对高通量测序数据进行预处理和分析,包括基因表达分析、变异检测等。在转录组学研究中,Samtools也可以用于处理和分析RNA-seq数据,如转录本拼接、差异表达分析等。此外,Samtools还常常与其他生物信息学工具和数据库进行整合,如Gene Ontology(GO)和Bioconductor等。
- 总结
Samtools作为一款强大的生物信息学软件,其开发团队不断对其进行更新和维护,使其在处理和分析高通量测序数据方面具有更高的效率和准确性。通过使用Samtools的排序、去重复和转化文件等功能,研究者们可以更加方便地处理和分析测序数据,为生物信息学研究提供有力的支持。同时,Samtools还具有广泛的应用场景和与其他工具的整合能力,使其成为生物信息学领域不可或缺的工具之一。
以上就是关于Samtools的详细介绍。希望这篇文章能够帮助你更好地了解这个工具在生物信息学中的应用。如果需要了解更多信息,可以查看其官方网站和相关教程进行学习。



















